

Matteo Chiara1,2, Graziano Pesole2,3
1Department of Biosciences, University of Milan
2Institute of Biomembrane Bioenergetics and Molecular Biotechnologies, National Research Council (IBIOM-CNR)
3Department of Biosciences, Biotechnologies and Biopharmaceutics, University of Bari “A. Moro”
Table of Contents
Last update: 2020/11/2017
- Introduction: from DNA to genomes (going through a PC)
- The assembly of a genome
- The annotation of a genome
- Gene banks
- Genomes and genomics for the control of pandemics
- Genomics and SARS-CoV-2: resources and applications
- Sharing information: “primary” data banks
- Processing the information: “secondary” resources [updated 2020/11/17]
- Limitations and challenges for the future: beyond genomes?
- Conclusions
For “non-experts”
References
Introduction: from DNA to genomes (going through a PC)
The capability to accurately determine the genetic heritage of an organism, which corresponds to the information present in its genome, has been a fundamental step for the development of modern molecular biology1,2. Nucleic acids, DNA and RNA, are macromolecular molecules constituted by long nucleotides chains and are the chemical components of the genomes. They differ for the sugar in the nucleotide – which is ribose for RNA and deoxyribose for DNA. Tha vast majority of genomes is made of DNA, typically in its double-stranded form of two complimentary filaments in which the nitrogen purine bases adenine and guanine match respectively with the nitrogen pyrimidine bases thymine/uracil and cytosine. However, some genomes are RNA-based.
The biological information, that is the universal code of life, is enclosed in the sequenze of nucleotides which may be of four types: adenine (A), cytosine (C), thymine (T; uracil (U) in RNA) and guanine (G). The knowledge of a genome sequence means to reveal pivotal information on the molecular mechanisms which regulate all the biological functions. For example, permits, through a process termed “annotation”, to identify and characterize the genes, that is the “pieces” of the genome which mediate and control the complex biological processes at the basis of the functioning of each cell type and, therefore, of the life of an entire organism.
According to the modern evolution theory, all the living organisms descend from a common ancestor, through a change (mutation) process and selection which would have started more than three billions of years ago3. Consequently, many living organisms, especially those strictly related, share a large part of their genes and show high levels of similarity in the sequence of their genomes. Taking advantage of this principle, through the direct sequence comparison of one or more genomes belonging to different organisms, the discipline named comparative genomics (comparison of genomic sequences) is able to retrieve essential information on the laws that rule genomes evolution and on the localization of the most important or indispensable sequence elements within a defined genome (e.g., the genes)4.
The same principle may be applied and extended to other research fields, such as genetics and medicine, where – for example – through the comparison of genomic sequenze of healthy and affected individuals – it is possible to identify genetic variants (e.g. portions of the genome which differ at the sequence level) associated to a specific pathological condition and, eventually, to determine their molecular function. Comparative genomics has been tremendously implemented in the past few years, thanks to the availability of an increasing number of complete genomic sequences. This led to an extraordinary revolution in the field of Life Sciences with the advent of the Genomics Era. The cause of this rapid transformation is mainly due to the development of new highthroughput sequencing methodologies, which are known as Next Generation Sequencing (NGS)5.
The first laboratory techniques for the determination of nucleic acid sequences were developed in the 70’s, in Frederick Sanger and Walter Gilbert labs (both recognized with the Nobel prize)1,2. Those methods were highly hard-working and had to be performed by highly specialized personnel. The yield was limited when compared to the modern sequencing techniques. It is estimated that in the earlt 80’s, with these sequencing methods, an individual could sequenced a genome constituted by 30000 DNA base pairs sequenziamento (the dimension of the SARS-CoV-2 genome), working without interruption for more than 60 days.
As for many other human processes, sequencing techniques have been constantly optimized and improved overtime. The increasing level of automation and the engineering of the sequencing process allowed the acquisition of ever higher production and accuracy levels. For a review of the history of the different sequencing technologies, of their respective mechanisms and of the production levels reached during the past years, please refer to the book Fondamenti di bioinformatica, edited by Zanichelli6. In this context, to have an idea of the advances acquired in the past 30 years, we have to think that the Human Genome Mapping Project (more than 3.000.000.000 DNA base pairs), which was launched in 1990, required 3 billions dollars and 15 years for completion. Today, by using the most advanced sequencing technologies, compared results may be obtained in few days, with a cost of about 1000 dollars7 (Figure 1).
Figure 2 Operative costs for the sequencing of a human genome.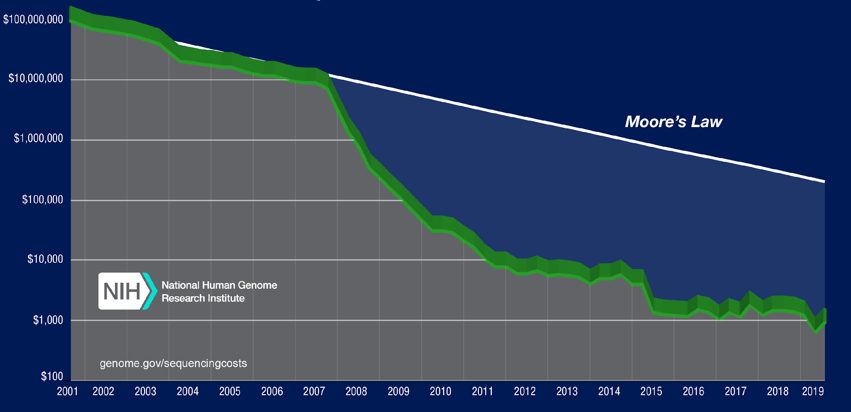
The picture shows the estimated costs (Y axis, log scale) for the sequencing of a human genome, in the past 20 years. The white line (Moore’s Law) shows the trend of the first Moore’s law, a hypothesis which describes the decrease in the cost of electronic components in the same time frame. Since the capability of the electronic components doubles (according to the Moore’s law) every 18 months, in the same period the cost halves (exponential decrease). Therefore, nucleic acid sequencing costs during the last years have been strongly reduced. [Source: NIH: genome.gov/sequencingcosts]
The reconstruction of a genome sequence is a complex process, which requires the generation, managing and analysis of a big amount of data. This concept applies also for relatively small genomes and was immediately evident since the first project for the sequencing of a genome in the late 70’s. In that period, researchers of professor Sanger’s group were involved in the reconstruction of the genome sequence of the bacteriophagus ϕX1748. The sequencing data were annotated on laboratory notebooks of 9 different researchers, each one emloyed in the analysis of a different genome segment. The reconstruction of the complete sequence required the integration of all the sequencing data, a problem which was not easy to solve, as the big amount of information, related to the 5000 base pairs of the viral genome, had to be linked in a congruent and codified manner. Fortunately, one of Sanger’s collaborator, doctor Michael Smith, had a brother-in-law, Duncan McCallum, who worked as a programmer in an informatic company in Cambridge. To facilitate the phage genome sequencing reconstruction, McCallum developed a computer software (in COBOL language, on perforated cards), which allowed to:
- Integrate the sequences produced by different researchers
- Reconstruct the complete genome sequence
- Look for specific sequences within the genome
- Translate in-silico the DNA sequences into proteins, thanks to the knowledge of the genetic code
The software was extremely useful to reconstruct the genome sequence of the phage ϕX174 and demonstrated how the application of a computer and of softwares developed ad-hoc to analyse biological sequences was indispensable and necessary for the study of genomes. Bioinformatics was born.
The success of the application of informatic methods for the study of biological sequences was so remarkable that in few years, in 1981, data banks dedicated to the collection of sequencing data (EMBL9 and Genbank10, which still constitute a reference point essential for all the biologists around the world) and tools specifically studied for the interrogation of sequencing data banks and the search of sequence similarities, such as the softwares FASTA11 and BLAST12, which are the most utilized tools by the scientif community worldwide, developed. During the last years, because of the constant increase of the amount of products, the development of softwares applications dedicated to the management of biological sequencing data analysis (but not only of them) became ever more pivotal for Biology and the discipline known as Bioinformatics became fundamental to reconstruct, analyse, decode the genome sequences. For a kore complete overview of themultiple applications of bioinformatics, please refer to Fondamenti di bioinformatica, Zanichelli6.
As mentioned before, the recent development of genomics (the discipline which study genomes) is strictly linked to the development of new sequencing technologies, more rapid and efficent. In the last 15 years, these technologies have revolutionised research in the molecular biology field. Research projects which would have required years of efforts and huge costs are now feasible in small time periods and with the employment of relatively small amount of personnel and money. To mark the discontinuity with the past the acronym NGS (Next Generation Sequencing) was coined to qualify these technologies. NGS technologies permit to perform more reactions in parallel, producing millions of sequences at the same time. The exponential increase of the amount of sequencing data generated by these advanced technologies, required a similar upgrade of the computational resources necessary to analyse data, as well as the development of new algorithms and softwares to make the analyses more treatable and efficient form a computational point of view. This process made bioinformatics and bioinformatic applications for the analysis of massive sequencing data more pivotal for biology.
All the molecular biology application took adavantage from the advent of NGS technologies. In fact, these applications range from the quantification of the mRNAs within a cell or tissue, to population genetics, to genomes reconstruction, to the study of transcription factors and chromatin architecture, and, more generally, to the study of regulatory elements of gene expression (such as enhancers, silencers), or even to the study of entire microorganisms communities (metagenomics). All these analyses may be performed in small time frames and with relatively low costs, producing an unprecedented amount of data13.
The assembly of a genome
Despite the new NGS technologies represented a great advantage, not all the problems related to the reconstruction of a defined sequence have been solved. One of the main limitations is that, even if sophisticated and except for very rare cases, no sequencing technology, at present, is able to provide an accurate, complete and unumbiguous representation of the genome sequence of an organism.
In fact, although many differences exist among the variety of available technologies, none of these is capable to read DNA sequences similar in lenght to that of a moderately complex organism. Furthermore, to date, the technologies able to reconstruct sequences with major lenghts, are typically associated with a high error rate5.
For this reason, as in the 70’s, the reconstruction of a complete genome sequence requires the use of dedicated softwares for the analysis of sequencing data, through a process known as assembly14.
The term assembly of a genome is referred to the procedure trough which the data obtained from a sequencing project (which represent discrete portions of a genome) are joined to form contiguous sequences of major dimensions, ideally corresponding to each DNA molecule (chromosomes, organelles genomes and plasmids) within the cell. The obtained sequences are called contigs. The assembly process is usually performed by specific computer softwares, called assemblers. The optimal result is constituted by a series of complete contigs which represnt in a 1:1 ratio each of the molecules present in the genome. However, since tha majority of genomes of complex organisms holds long repeated sequences, which cause problems during the assembly process, this result is particularly difficult to achieve.
The annotation of a genome
Once a genome sequence is reconstructed, the next step is to determine its functional elements, such as genes, promoters and other regulatory sequences (enhancers, silencers).
The process which recognizes the different functional elements of a genome is called “annotation”. the annotation of a genome is usually performed using appropriate bioinformatic tools and/or producing new sequencing data focused to the annotation of genes (e.g., the sequencing of transcription products, constituted by discrete RNAs)15.
The annotation based exclusively on the use of softwares is called in silico annotation of genomes. In fact, whenever possible, the comparison with a relatively similar genomic sequence already annotated, allows a rapid identification of the conserved functional elements (homologs). Alternatively, the functional elements of a genome may be predicted applying methods ab initio. Such methods take advantage of probabilistic algorithms to determine, given the properties of a nuclotide sequence (such as the composition or the presence of particular motifs), the probability that the sequence contains a gene or another functional element. The modern sequencing technologies allow to apply particular laboratory protocols which permit to sequence the intermediate products of the functional elements, that is transcripts. For example, it is possible to sequence, in a defined cellular type, messenger RNAs (mRNAs), small non coding regulatory RNAs (sncRNAs) and to obtain information on their coding regions simply mapping (i.e. searching identical genomic sequences) the obtained sequences on the reference genome of the same organism.)
All these approaches may be combined to obtained more complete information.
Gene banks
Once a genomic sequence has been reconstructed and annotated is made available to the scientific community through appropiate resources and data banks, which allow the navigation through the different annotations, the rapid comparison with other available sequences, the extraction of one or more similar sequences and the graphical visualization of the annotated genomic elements. Among the best known we may cite Genbank, a data bank of biological seqeunces of the NIH (National Institute of Health)10, one of the most renowned public insitution for medical research worldwide, or the sequences data bank ENA16, developed and maintained by ELIXIR, the european infrastructure for life sciences, or the genome browsers UCSC17 ed ENSEMBL18. These are advanced softwares tools which allows the navigation through the annotated functional elements in a genome thanks to an intuitive and simple graphical interface.
The public and free availability of data for all the scientific community, joined to the different tools that specialized data banks give to researchers is of pivotal importance for the constant improvement of our knowledge of the genomes and of the mechanisms which rules their evolution and use. Therefore, the development and the implementation of these resources and infrastructures represent a continuous and constant effort of bioinformaticians all around the world.
Genomes and genomics for the control of pandemics
Between the end of 2013 and the beginning of 2014, a lethal hemorragic fever – not diagnosed fro months – spread in southern Guinea (Guinée forestière). When the disease was diagnosed as Ebola, the virus already spread in three countries19, probably beyond the point into which local containment measures, such as isolation and the focused control of the infection chains, could have controlled the epidemic. In 2015, a new disease, similar to the Dengue fever, has been implicated in a dramatic increase of microcephaly cases in Brazil; one year later, the analyses revealed that the Zika virus spread in Americas, undetected by the surveillance systems, since the end of 201320.
These recent experiences demostrate how, despite the public health surveillance systems have evolved to satisfy the continuous demands of the population worldwide, we keep on drastically underestimate our vulnerability to pathogens, both old and new21. The recent events in western Africa and Brazil highlight gaps in the surveillance systems of infectious diseases, in particular when new pathogens, whose geographic area extended to a new region, have to be identified. Despite the lessons from the previous epidemics22, such as the severe acute respiratory syndrome (SARS) in 2002-2003 and the pandemic of influenza in 2009 – infectious events still surprise and, sometimes, overwhelm the control systems both at local and global level.
The implementation of monitoring systems of highly infectious and potentially lethal pathogens requires rapid and complete surveillance methods. The monitoring systems used at the moment are based on counting cases at local level and on simple techniques of partial sequencing (genotyping) to classify pathogens; these surveillance methods could be greatly improved through modern genomics.
The complete sequencing of the genome of pathogenic agents has been used for decades to understand viral epidemic transmission, since the first studies on hantaviruses in US23, to the human immunodeficiency virus (HIV) in the United Kingdom24; more recently, the approach has been successfully extended to pathogenic bacteria25. Genomic epidemiology, the discipline which use the sequencing approach of a pathogen for the study of epidemics, allows, at first, to identify the pathogenic agent and, thereafter, by the comparison among genomes, to determine both the evelutionary mechanisms of the genome of the pathogen and, thereafter, single transmission events within epidemics sites (contagion chains)25. More recently, this approach has been demostrated to be a crucial tool to rapidly answer to new infectious diseases hotspots26,27.
Many pathogens, especially viruses, mute their genome sequence vary fast. Consequently, isolated pathogens from strictly linked infection cases show sginificant differences at the genomic level and are clearly discernible.
These differences may be used to deduce conclusions from an epidemiological point of view and to reconstruct probable chains of contagion28. The complete sequencing of the genome of an emerging virus may provide a significant amount of information on the nature of the pathogen through the comparison of existing sequences. As long as a major number of sequences becomes available, the analysis of the genetic diversity among the population of the new pathogen may estimates the diffusion velocity and help to foresee the future development.
In the studies related to the chains of transmission, genetic variants are used to identify transmission events fron an individual to another one, through the analysis of genetic variants shared among pathogens isolated from different cases in a hotspot. In this way, it is possible to go back in detail the entire transmission network28. Highthroughput genomic investigations related to the diffusion of a pathogen, such as epidemics, are very different. Given the elevated level of diffusion of the disease, only a subset of the genomes of the pathogen may be isolated and sequenced. Therefore, the goal is to use genomic sequencing data to understand the epidemic general dynamics and the possible evolutionary mechanisms of the pathogen. This approach, integrating the principles of epidemiology and evolutionary biology, is termed phylodynamics29. Since the large part of the pathogens, and in particular single-stranded DNA viruses, RNA viruses and many bacteria species, mutate rapidly, through phylodynamics is possible to cobine epidemiological observations with evolutionary analyses, to unveil how the dynamics which rule the evolution of the genome of a pathogen may impact on the clinical manifestation of the disease30.
Genomics and SARS-CoV-2: resources and applications
The recent application of genomics to the monitoring of COVID-19 pandemic highlighted the importance of fast sequencing genomic-based approaches for a really serious public health crisis.
Between 2019 and 2020, a group of scientists, applied a particular sequencing technology, which allows to determine the RNA sequence of all the living species in a biological/environmental sample (metagenomic sequencing of total RNA) to analyse the broncho alveolar lavage fluid (BALF) obtained from a single patients in Wuhan, Cina, where many cases of severe respiratory infections were reported31. Thanks to the application of bioinformatic tools, Wu and collegues reconstructed genomic sequencing data of the potential pathogenic agent, a new coronavirus thereafter named SARS-CoV-2. During the first days of 2020, the complete sequence of the viral genome has been doposited in data bank and made available to the entire scientific community. The identification of the genomic sequence of the new pathogen facilitated the development of rapid molecular diagnostic tests all over the world32. These tests have been fundamental to monitor and contain the spread of the disease. Unfortunately, SARS-CoV-2, the etiological agent of COVID-19 pandemic spread very rapidly at the global level. Once again,the techniques based on the reconstruction of the sequence of the genome have been the keystone of a worldwide and coordinated effort to study the transmission of the disease and the evolution of the virus both at national and international levels.
Examples are the SPHERES project33 (SARS-CoV-2 Sequencing for Public Health Emergency Response, Epidemiology and Surveillance) in the US or the COG-UK project34(COVID-19 Genomics UK ) in the UK. This latter, launched in march 2020, is probably the most complete monitoring system of COVID-19 pandemic. COG-UK consists in a capillary network of genomic surveillance at the national level which aims to trace SARS-CoV-2 transmission, identify new mutations in the genome and integrate genomic data with epidemiological and health data. To date, (August 14th, 2020), COG-UK has identified more than 35.000 SARS-CoV-2genomes. These data have been crucial to reconstruct in an accurate manner the transmission dynamics of COVID-19 in the United kingdom. On the basis of SPHERES and COG-UK, similar initiatives have been implemented at the national level in those countries affected COVID-1935,36,37,38. The availability of this huge amount of genomic data, in real time, has been fundamental to trace the chains of contagion and to oppose an informed and organized resistance to SARS-CoV-2 spread.
Sharing information: “primary” data banks
The GISAID data bank (Global Initiative on Sharing All Influenza Data) is, at present, the landmark at the global level for genomic data sharing of viral pathogens and the study of emerging mutations which may be relevant for the control of outbreaks and vaccine development39. The initiative, which led to GISAID birth, has its roots in 2006, when the hesitancy to share the genomic sequences of the avian influenza H5N1 virus, created a global emergency, uncovering some dangerous deficiencies and limitations in the GINS (Global Influenza Surveillance Network) surveillance system of the World Health Organization (WHO)40. On the basis of the acquired experience, the scientific community elaborated new principles and rules to promote international sharing of alla the data related to influenza viruses and to public results in a collaborative manner. Initially, this was simply an expression of intent, but later became an indispensable mechanism to favour, through data sharing, a more coordinated and efficient reaction to flu outbreaks.
The EpiFlu database, the GISAID resource for genomic data sharing of influenza viruses has been pivotal for the answer to the influenza A (H1N1) pandemic in 2009, allowing to promptly monitor the evolution of the new virus while spreading worldwide41. Thereafter, in 2013, the availability of the genomic sequences of H7N9 virus has been of great importance to generate, develop and test vaccines, through synthetic biology approaches, within few weeks from the isolation of the virus42.
To date, EpiCov web resource, the GISAID tool developed to specifically collect COVID-19 pandemic data, represents the most used and complete archive of SARS-CoV-2 genomic data. EpiCoV provides a collection of more than 80000 complete genomes, isolated from more than 80 countries (data collected on August, 14th, 2020). Besides the genomes, EpiCoV makes available a variety of metadata – although limited – including the sample type from which the virus has been characterized, the sequencing technology and applied protocols, but also basic clinical annotation, that is patient clinical conditions (hospitalized or asymptomatic), sex, age. Moreover, EpiCoV provides weekly the processing of genomic data to identify the most recurrent and diffuse mutations in the viral genome and possible mutations with a potential impact for the clinics and diagnostics. Despite EpiCov data are public, the access is reserved only to registered users, the data connot be re-distributed and their use is limited to only research aims. Finally, not-processed sequencing data (not assembled sequences) cannot be deposited.
For these reasons, the Research Data Alliance (RDA)43, a group of scientists which is involved to define the optimal fundamentals for scientific data sharing, elaborated a series of recommendations and guidelines, to facilitate a major sharing of data obtained from SARS-CoV-2 sequencing. In particular, RDA strongly suggest that SARS-CoV-2 genomic data have to be available through resources with controlled access such as GISAID, but also that the primary sequencing data, as obtained by the sequencing machine, should be deposited also in data banks with free access, such as the web resource for viral genomes developed by Genbank10 or the analog portal for the collection of SARS-CoV-2 genomes, developed by the european data bank ENA16. The free access to primary sequencing data not yet elaborated is pivotal to grant the complete transparency and reproducibility of the analyses performed by different reesearch groups. Moreover, their aggregation may allow, immediately or in the future, the execution of new analyses, which, through the application of improved or different bioinformatic tools, could retrieve novel information on the evolutionary mechanisms of the virus or on the pathogenesis of the disease.
Processing the information: “secondary” resources
To answer to COVID-19 pandemic an unprecedented amount of data has been generated.
To navigate into this enormous quantity of data to find the information of interest, may be a process not always easy and immediate. For this reason, the scientific community rapidly implemented computational infrastructures to facilitate the access and recovery of different kind of COVID-19-associated data.
The COVID-19 portal (www.covid19dataportal.org) of the European Bioinformatics Institute (EBI) and the twin portal developed by the NIH (www.ncbi.nlm.nih.gov/sars-cov-2) are, probably, the resources which, actually, provide the most complete catalogue to navigate, access and obtained SARS-CoV-2 sequencing data and the best bioinformatic tools to use for their analysis. The portal protocols.io (www.protocols.io), harvest a collection of more than 150 laboratory protocols and in silico (bioinformatic), for the generation, manipulation, analysis and deposition in data banks of SARS-CoV-2 data. In parallel, a detailed list of laboratory protocols, bioinformatic methods and data bankds which contain SARS-CoV-2 sequencing data has been made available by Github resource, of the US Centre for the Control and Prevention of Diseases (CDC, github.com/CDCgov/SARS-CoV-2_Sequencing). Similar initiatives have been also developed at the national level, such as the COG-UK (www.cogconsortium.uk) or the COVID-19 Data Portal Sweden (www.covid19dataportal.se), developed by swedish researchers.
Besides the web resources for data sharing, the scientific community developed different softwares and strategies to overcome the challenge of their integration and interpretation. Application and easy-to-use methods for SARS-CoV-2 strains classification, based on the direct comparison of complete genomes through phylogenetic trees and on more robust stratification strategies, which use only those more frequent genomic variants, have been generated44,45,46. To ease the interepretation of these result, dedicated web tools ahve been created, to visualize in an interactive way the circulating viral strains in different areas of our planet (e.g., Microreact e Nextstrain47,48; Figure 2).
Figure 2 Systems for the visualization of phylogeographic data.
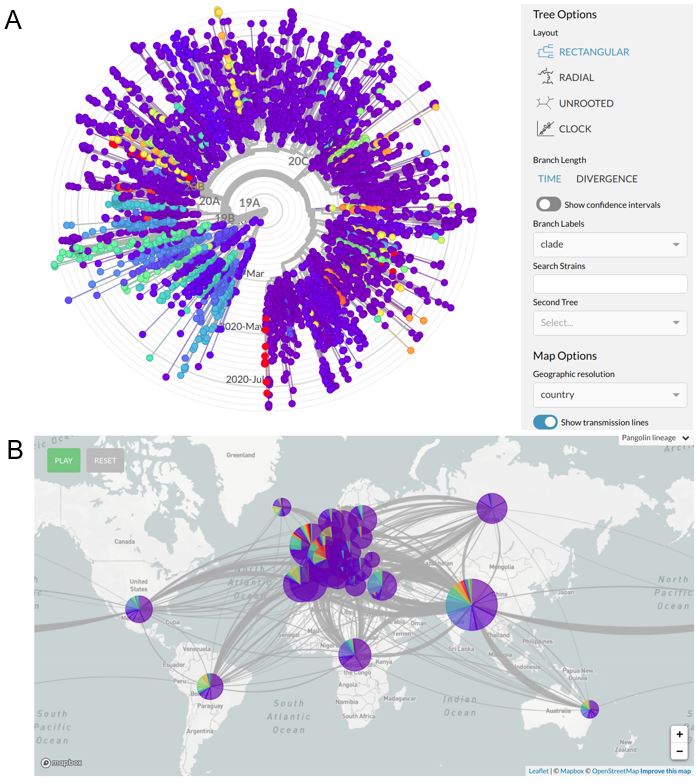
A A phylogenetic tree is a type of graphical representation of the gentic distances among organisms, which allows to visualize the most similar entities. In the Nexstrain platform, the phylogenetic trees may be used to visualized the relationships among SARS-CoV-2 isolate. Drop-down menù allow different levles of configuration. B Detailed map of the connection among different viral strains circulating all around the world, in real time with viral transmission dynamics. [From: nextstrain.org]
Taking advantage of the genomic sequences available in GISAID, Korber and collegues49 developed a series of dedicated binformatic tools to trace changes in the SARS-CoV-2 Spike glycoprotein, which mediates the entry of the virus in the host cell and which represent one of the best target for the development of a vaccine. Integrating the genomic analyses with clinical data the authors have found a particular aminoiacidic substitution in the Spike protein sequence (D614G; D = aspartic acid; G = glycine) which seems to correlate to potentially elevated viral loads and which may be the result of an adaptation process of the virus to the human host. However, to date, the clinical implications of this observation which, in the lack of experimental validation, is simply a valid speculation, are still not clear and also according to the authors the presence of this variants does not seem to associate to a more unfavourable prognosis. Similar observations, which have to be still validated, have been reported also by other authors and constitute a good starting point for the execution of focused analyses to understand the molecular mechanisms at the basis of COVID-19 pathogenesis.
[Update 2020/11/17] It has been finally established that the D614G variant is effectively able to more efficiently infect cells of the upper respiratory and lungs64. The G614 variant replicates more efficiently in the upper airway cells, leading to high viral loads, but not in the lung. Therfore, as mentioned in other works, the G614 variant does not correlate with a more severe disease. In hamsters previously infected with the D614 variant a more potent immune response is elicited against the heterologous G614 strain. This means that vaccines undergoing current phase III clinical trials are still the goal to achieve to defeat this pandemic. On the other hand, the recognition ability of monoclonal antibodies, another high hope for COVID-19 therapy, has to be carefully tested. Indeed, all the synthetic antibodies produced to date have been generated taking into account the D614 viral type. The G in position 614 Spike seems to confer conformational changes to the protein, limiting the recognition by some monoclonal antibodies65. [end of the update]
Limitations and challenges for the future: beyond genomes?
The large part of pathogens surveillance systems integrates different kind of data, including those from genomics, to perform the surveillance of a specific disease. The pathologic condition of interest is monitored at different levels, through diagnostic tests, epidemiological models, genomic data or, in case of emerging hotspots, rapid notification of unexpected sistuations. Despite the remarkable progresses reached in the previous decades, more groups of experts have continously underly the requirement of a better surveillance capacity of infectious diseases50. An ideal global surveillance system should be based on a more agnostic approach and not pathogen-specific51. Such a surveillance system should take advantage and integrate new data, like “big data”, which are made available by novel digital technologies. For example, monitorng different fluxes of data, such as school truancy or absenteeism from work places, the purchase of specific food or medicines, or the flux of patients into hospitals, or the automated analysis of leaning words or sentences on the social media such as Twitter52 or in searches in Internet53.
This new surveillance approach, known as digital epidemiology, could be very useful for the digital monitoring, in real time, of infectious diseases54. Digital epidemiology first recovers information from a series of sources, including digital media, news reports, official bulletins and “crowdsourcing”; therafter, relevant epidemiological data are extracted and elaborated in a series of reports, which are analyzed to identify possible critical situations. At the moment, at least 50 digital epidemiology platforms are operative all around the world54.
Despite these platforms are not yet integrated in the official monitoring systems established by the scientifc community, their flexible nature and the possibility to obtain real time data make these platforms efficient tools to recover information regarding an outbreak, in particular in those contexts lacking traditional systems of disease surveillance.
Recent experiences with Ebola and Zika viruses have domostrated how digital epidemiology domains anf genomics could be integrated in the future to offer a more detailed and clear overview of an ongoing outbreak. During Ebola outbreak monitoring in west Africa, digital epidemiology-based approaches have revealed that interface places between rural and urban populations constituted one of the favourite turning point for viral diffusion55, echoed by the results obtained from a genomic study related to the main contagion chains19. During the Zika outbreak in Brazil, Majumder and collegues used Google Trends to estimate the basal reproductive number -R0- of the virus56 obtaining a value analog to that retrieved by phylodynamic analyses and genomic data (1.29-3.85)20. These results indicate that both the types of approaches, phylodynamics and digital epidemiology, may be used for the calculation of fundamental epidemiological parameters which help to understand virus diffusion and to organize an appropriate response for the safeguard of public health.
In a near future, it is not difficult to imagine a complete integration of genomic and digital epidemiological data57. The potential of such an epidemiological surveillance system would be higher than the potential of the current systems. Digital epidemiological data could be used to address reasearchers to those regions in the world in which epidemiological critical situations are potentially ongoing. Threafter, in these regions systematic sequencing approaches of samples collected from infected wild animal species, or human patients, could be implemented to identify new pathogens. Genomic sequencing data could be, in turn, integrated in real time with ever more rich metadata and made available through dedicated web platform, such Microreact for the collaborative analyses of sequencing data and Nextstrain to viualize results. These resources – or similar resources – already used to respond to Ebola and Zika outbreaks or to COVID-19 pandemic, should be a point of aggregation for a global network of experts, contributing to epidemiological and phylodynamic analyses, allowing the identification of possible spillover events, the expansion of a specific pathogen and/or the transmission of a new infectious agent among humans. The results would be immediately shared with the personnel at the forefront in fighting infectious diseases – epidemiologists, veterinarians and healthcare workers – allowing to adopt rapid and focused containement measures to mitigate the futher spread.
Conclusions
The adaptation of a new pathogen to the human species – the so-called leap-of-species events or spillover – are not novel across the human history, although, fortunately, events like COVID-19 pandemic are exceptional58. The possiblity to share more and more inforamtion, to analyse data through better and more efficient algorithms and to promptly obtained rapid and complete answers to complex problems, is, for sure, one of the main weapons which, in the future will allow the human being to prepare more capillary newtworks to control and circumscribe possible emergencies, such as outbreaks caused by the onset of new pathogens59. It’s easy to foresee that bioinformatics and genomics will play more central roles in the implementation and setting of monitoring systems, demontrated also by the prompt and efficient response that these disciplines have given to counteract the current emergency situation.
The efforts produced for SARS-CoV-2 genomic data generation and analysis is unprecedented in the history of modern Science. In fact, the production of such an amount of data in a very small time frame would have been inconceivable only few years ago. However, to elaborate even better strategies, in the future the decisional process will have to be rapid and provided with accurate and real time data and, ideally, integrate different levels of inofrmation, including genetic and molecular data of both the pathogen and the host, epidemiological and demographic data and, whenever possible, the health conditions and the clinical history of the patients.
Despite the answer of the scientific community to the Covid-19 pandemic has been incredibly rapid and efficient, some critical issues, which have to be solved in the near future, are reported.
In this regard, it is clear that the standardization of protocols and methods used to generate and analyze data and the development of integrated resources to allow a fast and constant access to all the available information, constitutes one of the most relevant challenges for the future of modern biology. In fact, whereas many databases and resources are currently available to privide the access to SARS-CoV-2 sequencing data, the integration of all the data and metadata stored in these archives is not always simple and limits possible future applications and studies. For example, according to our analyses, because of some inconsistency in data deposition systems or annotation errors, all the main data banks of SARS-CoV-2 genomic sequences show a certain degree of redundancy and contain equivalent and overlapping information which are not easily discernible. In the absence of certain and defined rules for data deposition and metadata annotation, it is basically impossible to precisely understand how the information and data are duplicated in different archives. This phenomenon complicates the integration of available data and has pronouced implications for the execution of statistics studies which may allow the identification of genetic variants in the host or in the virus, eventually associated to a more aggressive disease. Moreover, despite almost all SARS-CoV-2 genomes come from clinical institutes, frequently patients data are not made available to the scientific community, even in an aggregated form. This consistently limits the type of analyses which may be performed. Similar considerations may apply to clinical trials for the evaluation of vaccine efficacy and other therapeutic approaches.
Genetic association studies60 have been demonstrated fundamental in different fields to understand the characteristics of our genome. It is reasonable to hypothesize that whether statistic samples of adequate dimension were available, these studies could be useful to better understand SARS-CoV-2 physiology and the different COVID-19 clinical manifestations, with possible applications also in the design of new vaccines and therapeutic routes. Nevertheless, despite some noteworthy initiatives61,62,63, currently the execution of genome wide association studies for COVID-19 is limited, probably also because of the complexity in obtaining and integrate all the required data and metadata. Although the human being has made excellent progresses from a technological and scientific point of view, the COVID-19 pandemic has highlighted alarming criticisms, also and especially in the communication systems and in the management of information64.
In fact, also in our Country, incomplete information and notions, not precise or simply false have too easy circulated through different communication channels, loading the population with unwarranted fears and risks. For this reason, we hope that in the future the pandemics monitoring systems may be integrated not only to incorporate higher number of data but also to allow a more accurate and correct communication of the relevant information to the population. Such a process requires also a little cultural revolution, for an appropriate use of communication and social media and necessarily goes through the promotion of a more seroious and diffuse scientific culture to the population (but also to the heads of media). A process that, thanks to the modern communication instruments, could be articulated also through more inclusive and capillary citizen science initiatives, with the direct participation of citizens to big research projects (e.g., through the use of mobile phone apps which may communicate critical information for the monitoring of pandemic episodes, such as the onset of fever or other symptoms). Through this new paradigm, which involve more directly the citizens in research projects – e.g., making them an active part in the collection, processing and in some steps of data analysis – it could be possible making citizens more suited to the correct use of the scientific method, avoiding the spread of groundeless information, negationisms and other different kind of amenities which are often conveyed in an incorrect manner by social media.
For “non-experts”
Silencer
A region in the genome which has the possibility to switch-off the expression of a gene.
Enhancer
A region in the genome which has the possibility to potentiate the expression of a gene.
Promotor
A region in the genome from which the expression of a gene starts.
Organelle
A compartment within the cell with specialized function (e.g., the mitochondrion, which is the “respiratory” organelle and represent the source of energy for the cell).
Metadata
Bioinformatic metadata are a group of information which describe a series of data (e.g., where, how and when sample related to specific data has been collected, which where the environmental conditions etc.)
References
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 1977; 74:5463–5467.
- Maxam AM, Gilbert W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. 1977; 74:560–564.
- Weiss MC, Preiner M, Xavier JC, et al. The last universal common ancestor between ancient Earth chemistry and the onset of genetics. PLOS Genet. 2018; 14:e1007518.
- Alföldi J, Lindblad-Toh K. Comparative genomics as a tool to understand evolution and disease. Genome Res. 2013; 23:1063–1068.
- Koboldt DC, Steinberg KM, Larson DE, et al. The next-generation sequencing revolution and its impact on genomics. Cell 2013; 155:27–38.
- Citterich MH, Ferrè F, Pavesi G, Pesole P, Romualdo C. Fondamenti di bioinformatica, Zanichelli, 2018.
- The Cost of Sequencing a Human Genome. Genome.gov.
- Sanger F, Coulson AR, Friedmann T, et al. The nucleotide sequence of bacteriophage φX174. J. Mol. Biol. 1978; 125:225–246.
- Kanz C, Aldebert P, Althorpe N, et al. The EMBL Nucleotide Sequence Database. Nucleic Acids Res. 2005; 33:D29–D33.
- Benson DA, Karsch-Mizrachi I, Lipman DJ, et al. GenBank. Nucleic Acids Res. 2007; 35:D21–D25.
- Lipman D, Pearson W. Rapid and sensitive protein similarity searches. Science 1985; 227:1435–1441.
- Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410.
- Applications of next-generation sequencing.
- Simpson JT, Pop M. The Theory and Practice of Genome Sequence Assembly. Annu. Rev. Genomics Hum. Genet. 2015; 16:153–172.
- Yandell M, Ence D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012; 13:329–342.
- Leinonen R, Akhtar R, Birney E, et al. The European Nucleotide Archive. Nucleic Acids Res. 2011; 39:D28–D31.
- Karolchik D, Hinrichs AS, Kent WJ. The UCSC Genome Browser. Curr. Protoc. Hum. Genet. Editor. Board Jonathan Haines Al 2011; CHAPTER:Unit18.6.
- Ensembl 2018.
- Dudas G, Carvalho LM, Bedford T, et al. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 2017; 544:309–315.
- Faria NR, Azevedo R do S da S, Kraemer MUG, et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016; 352:345–349.
- Gostin LO. Our Shared Vulnerability to Dangerous Pathogens. Med. Law Rev. 2017; 25:185–199
- Braden CR, Dowell SF, Jernigan DB, et al. Progress in global surveillance and response capacity 10 years after severe acute respiratory syndrome. Emerg. Infect. Dis. 2013; 19:864–869
- Nichol ST, Spiropoulou CF, Morzunov S, et al. Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science 1993; 262:914–917
- Holmes EC, Zhang LQ, Robertson P, et al. The molecular epidemiology of human immunodeficiency virus type 1 in Edinburgh. J. Infect. Dis. 1995; 171:45–53
- Popovich KJ, Snitkin ES. Whole Genome Sequencing-Implications for Infection Prevention and Outbreak Investigations. Curr. Infect. Dis. Rep. 2017; 19:15
- Kamelian K, Montoya V, Olmstead A, et al. Phylogenetic surveillance of travel-related Zika virus infections through whole-genome sequencing methods. Sci. Rep. 2019; 9:16433
- Kugelman JR, Wiley MR, Mate S, et al. Monitoring of Ebola Virus Makona Evolution through Establishment of Advanced Genomic Capability in Liberia. Emerg. Infect. Dis. 2015; 21:1135–1143
- Guthrie JL, Gardy JL. A brief primer on genomic epidemiology: lessons learned from Mycobacterium tuberculosis. Ann. N. Y. Acad. Sci. 2017; 1388:59–77
- Grenfell BT, Pybus OG, Gog JR, et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 2004; 303:327–332
- Drummond AJ, Suchard MA, Xie D, et al. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012; 29:1969–1973
- Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020; 579:265–269
- Carter LJ, Garner LV, Smoot JW, et al. Assay Techniques and Test Development for COVID-19 Diagnosis. ACS Cent. Sci. 2020; 6:591–605
- CDC. Coronavirus Disease 2019 (COVID-19). Cent. Dis. Control Prev. 2020;
- Data – COG-UK Consortium.
- SeqCOVID – Genomic epidemiology of SARS-CoV-2 in Spain.
- Gudbjartsson DF, Helgason A, Jonsson H, et al. Early Spread of SARS-Cov-2 in the Icelandic Population. medRxiv 2020; 2020.03.26.20044446
- Walker A, Houwaart T, Wienemann T, et al. Genetic structure of SARS-CoV-2 reflects clonal superspreading and multiple independent introduction events, North-Rhine Westphalia, Germany, February and March 2020. Euro Surveill. Bull. Eur. Sur Mal. Transm. Eur. Commun. Dis. Bull. 2020; 25:
- Genomics of Indian SARS-CoV-2: Implications in genetic diversity, possible origin and spread of virus | medRxiv.
- Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data – from vision to reality. Eurosurveillance 2017; 22:30494https://dx.doi.org/10.2807%2F1560-7917.ES.2017.22.13.30494.
- Butler D. Flu researchers slam US agency for hoarding data. Nature 2005; 437:458–459.
- Butler D. Swine flu goes global. Nature 2009; 458:1082–1083.
- The fight against bird flu. Nature 2013; 496:397.
- Research Data Alliance. Final release: COVID-19 guidelines. 2020.
- Chiara M, Horner DS, Gissi C, et al. Comparative genomics provides an operational classification system and reveals early emergence and biased spatio-temporal distribution of SARS-CoV-2. bioRxiv 2020; 2020.06.26.172924.
- Rambaut A, Holmes EC, Hill V, et al. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. bioRxiv 2020; 2020.04.17.046086.
- Galaxy and HyPhy developments teams, Nekrutenko A, Kosakovsky Pond SL. No more business as usual: agile and effective responses to emerging pathogen threats require open data and open analytics. 2020.
- Hadfield J, Megill C, Bell SM, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinforma. Oxf. Engl. 2018; 34:4121–4123.
- Argimón S, Abudahab K, Goater RJE, et al. Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microb. Genomics 2016.
- Korber B, Fischer WM, Gnanakaran S, et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020.
- Commission on a Global Health Risk Framework for the Future, National Academy of Medicine, Secretariat. The Neglected Dimension of Global Security: A Framework to Counter Infectious Disease Crises. 2016.
- Mandl KD, Overhage JM, Wagner MM, et al. Implementing Syndromic Surveillance: A Practical Guide Informed by the Early Experience. J. Am. Med. Inform. Assoc. JAMIA 2004; 11:141–150
- Aslam AA, Tsou M-H, Spitzberg BH, et al. The reliability of tweets as a supplementary method of seasonal influenza surveillance. J. Med. Internet Res. 2014; 16:e250.
- Hulth A, Rydevik G, Linde A. Web queries as a source for syndromic surveillance. PloS One 2009; 4:e4378.
- Brownstein JS, Freifeld CC, Madoff LC. Digital disease detection–harnessing the Web for public health surveillance. N. Engl. J. Med. 2009; 360:2153–2155, 2157.
- Zinszer K, Morrison K, Verma A, et al. Spatial Determinants of Ebola Virus Disease Risk for the West African Epidemic. PLOS Curr. Outbreaks 2017.
- Majumder MS, Santillana M, Mekaru SR, et al. Utilizing Nontraditional Data Sources for Near Real-Time Estimation of Transmission Dynamics During the 2015-2016 Colombian Zika Virus Disease Outbreak. JMIR Public Health Surveill. 2016.
- Khoury MJ, Iademarco MF, Riley WT. Precision Public Health for the Era of Precision Medicine. Am. J. Prev. Med. 2016; 50:398–401.
- Kolbert E. Pandemics and the Shape of Human History. New Yorker.
- Yozwiak NL, Schaffner SF, Sabeti PC. Data sharing: Make outbreak research open access. Nature 2015; 518:477–479.
- Tam V, Patel N, Turcotte M, et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019; 20:467–484.
- The COVID-19 Host Genetics Initiative. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 2020; 28:715–718.
- Ellinghaus D, Degenhardt F, Bujanda L, et al. The ABO blood group locus and a chromosome 3 gene cluster associate with SARS-CoV-2 respiratory failure in an Italian-Spanish genome-wide association analysis. medRxiv 2020; 2020.05.31.20114991.
- Ellinghaus D, Degenhardt F, Bujanda L, et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020.
- Al-Zaman MS. COVID-19-related Fake News in Social Media. medRxiv 2020; 2020.07.06.20147066.
- [update 17/11/2020] Plante, J. A. et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 1–9 (2020) [end of the update]
 Scarica il PDF dell'articolo
Scarica il PDF dell'articolo
Commenti